alphavirus.org
The McInerney Laboratory at Karolinska Institutet, Stockholm, Sweden
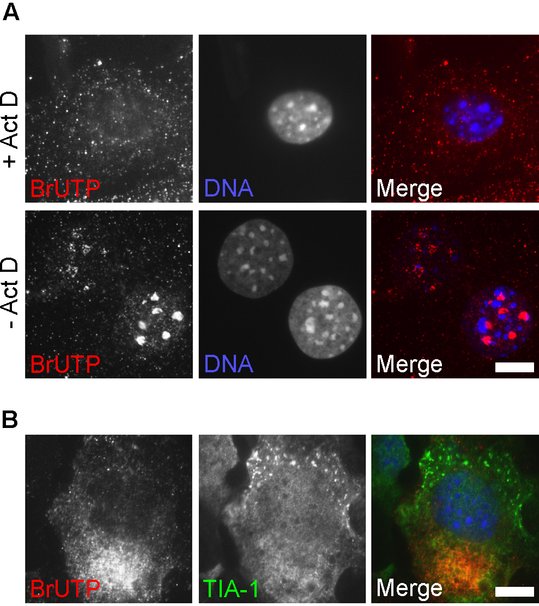
In vivo labeling of nascent viral RNA with BrUTP
Text and figure adapted from (Panas et al., 2015)
Since the function of SGs appears linked to the triage of mRNA, it is reasonable to assume that SGs induced during viral infection might sequester viral mRNAs in order to inhibit their translation. To examine the relationship between viral RNA replication complexes and SGs, it is useful to visualize nascent viral RNA in situ and co-stain for SG marker(s). A number of techniques are available for co-staining RNA and protein including 5-bromourudine 5’-triphosphate (BrUTP) labeling. BrUTP labeling involves incubating cells with modified ribonucleotide BrUTP to allow its incorporation into nascent RNA, followed by the immunodetection of BrUTP using a specific antibody (Enzo) (Restrepo-Hartwig and Ahlquist, 1996). BrUTP uptake into cells is facilitated by transfection. This technique allows the detection of viral RNAs, which may be localized to SGs during infection. Its major advantage over conventional in situ hybridization and dsRNA staining, if used together with actinomycin D (Act D; see below) to block host cellular transcription, it is selective for newly synthesized viral RNA produced during short times during infection. In our previous work (McInerney et al., 2005), we used in vivo BrUTP labeling to identify nascent SFV RNA in combination with TIA-1 immunostaining to identify SGs. We found that, when present in virus-infected cells, the SGs were typically located in regions of the cell devoid of newly produced viral RNA. As the infection progressed, and viral replication complexes were distributed throughout the cytoplasm, SGs were no longer detectable. We did not detect BrUTP labelled viral RNA in SGs, in agreement with FISH results that did not detect viral RNA in SGs.
Alphavirus infection profoundly inhibits the synthesis of cellular RNA (Gorchakov et al., 2005). Short times after infection, pulse-labeled RNA will consist almost entirely of viral RNA (Breakwell et al., 2007; Gorchakov et al., 2005). However, for other RNA viruses, BrUTP staining may label both viral and cellular RNA, confounding interpretation of staining. Since viral RNA-dependent RNA polymerases are not sensitive to actinomycin D (Act D), this inhibitor can be used during BrUTP labeling to ensure that only viral RNAs incorporate the BrUTP. Act D inhibits DNA-dependent transcription by binding DNA template and preventing mRNA elongation by RNA polymerases, but does not bind RNA. Figure A shows MEFs after staining with BrUTP for 1 hour in the presence or absence of Act D. In non Act D-treated cells, BrUTP staining is brightest in the nucleoli, the sites of ribosomal RNA synthesis. Different intensities of BrUTP staining reflect different efficiencies of transfection of the nucleotide in individual cells.
Protocol:
Grow cells on coverslips in 12-well plates and perform infection experiment so that cells are at approximately 70-90% confluency at time of fixation.
Optional: 30 minutes before each labelling period, medium is replaced with complete medium containing 1μg/mL Act D. Note that Act D is often difficult to solubilize; Actinomycin D-mannitol from Sigma (A5156) is formulated to avoid this problem and is water soluble.
Medium is removed and replaced with 450 μL OptiMEM (Invitrogen).
Prepare a mixture containing 45 μL OptiMEM, 10mM BrUTP (Sigma B7166) and 5 μL Lipofectamine 2000 reagent (Invitrogen). Incubate at RT for 15 min and pipette slowly onto cells.
Incubate cells for desired labelling time at 37°C.
Wash cells three times in ice-cold PBS.
Fix for 15 minutes in 4% PFA in PBS at room temp with shaking.
Permeabilize in MeOH/Triton as appropriate.
Stain with BrUTP-specific antibody (for example Enzo (ADI-MSA-200-E).
We infected MEFs with wt SFV at MOI 0.5, and labeled total RNA using a 1-hr incubation with BrUTP in the presence of Act D (between 4 and 5 hpi; Figure B). Under these conditions, no RNA staining was detected in nucleoli. In cells where SGs were visible, nascent viral RNA staining was always located contralateral to the TIA-1-positive SGs. A representative image is shown.
References
Breakwell, L., P. Dosenovic, G.B. Karlsson Hedestam, M. D'Amato, P. Liljeström, J. Fazakerley, and G.M. McInerney. 2007. Semliki Forest virus nonstructural protein 2 is involved in suppression of the type I interferon response. J Virol. 81:8677-8684.
Gorchakov, R., E. Frolova, and I. Frolov. 2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J Virol. 79:9397-9409.
McInerney, G.M., N.L. Kedersha, R.J. Kaufman, P. Anderson, and P. Liljeström. 2005. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol Biol Cell. 16:3753-3763.
Panas, M.D., N. Kedersha, and G.M. McInerney. 2015. Methods for the characterization of stress granules in virus infected cells. Methods. 90:57-64.
Restrepo-Hartwig, M.A., and P. Ahlquist. 1996. Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J Virol. 70:8908-8916.